

Abstract
Purpose
Acute lymphoblastic leukemia (ALL) is the most common cancer in childhood and has a high treatment success rate. However, survivors of childhood ALL have a high prevalence of chronic medical illnesses, such as metabolic syndrome. Dysbiosis in the gut microbiome is associated with metabolic derangement and disease risk. Chemotherapy and antibiotics increase gut microbiome dysbiosis. We tested the hypothesis that ALL treatment can cause alterations in the gut microbiome that persist in survivorship.
Method
Stool samples were collected on fecal occult blood test cards from 38 survivors between 2-18 years of age who were >1 year off therapy for ALL. 16 stool samples from healthy siblings age 2-18 years were collected from 14 families as controls. Clinical data including anthropometrics, antibiotic use, and probiotic use were collected. DNA was extracted and sequenced using a modified Illumina 16S Metagenomics Sequencing Library Preparation protocol for analysis of hypervariable region V4. A single rarefaction was performed at 4,897 removing 2 samples with low mapped read counts. Alpha and beta (Bray) diversities were calculated; Wilcoxon rank-sum test was used to compare the alpha diversities between groups, as well as the beta diversities within and between groups. Wilcoxon signed-rank test was used to compare the alpha diversity between survivors and their matched siblings. Finally, read counts for each Operational Taxonomic Unit (OTU) was normalized and tested for differential abundance using edgeR, assuming a negative binomial model.
Results
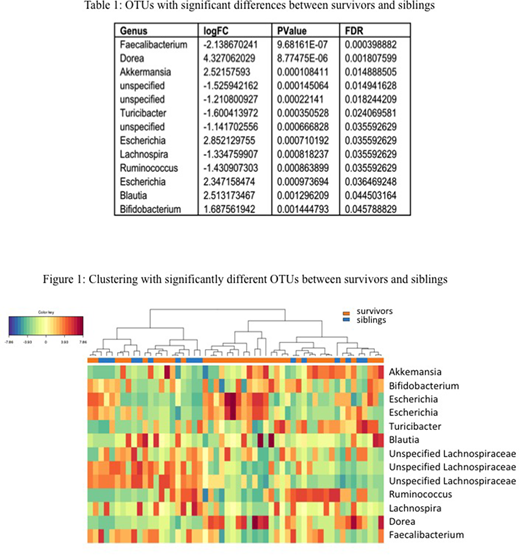
Significant differences in the abundance of taxa were found, with the most notable being the depletion of Faecalibacterium in survivors (FDR= 0.0004). Figure 1 shows a heatmap with the complete linkage clustering method on the Euclidean distance using the 13 OTUs with statistically significant (FDR<0.05) differences between survivors and siblings (Table 1). When compared to their respective siblings, survivors exhibited decreased alpha diversity using four metrics (Observed, Shannon, Simpson, Fisher), but these results did not meet statistical significance. Significant differences were not observed in alpha diversity within survivors between different clinical categories (antibiotic usage, probiotic usage, disease risk, age at diagnosis and time off treatment). Stool microbiome was more similar within a family than between families (P = 0.00079).
Conclusion
Differences in the relative abundance of certain taxa were found between survivors and siblings. Specifically, Fecalibacterium was depleted in survivors, which has also been previously observed in older populations of childhood ALL survivors. This study shows that these microbiome changes are present in ALL survivors during childhood, and should be evaluated longitudinally for association with the chronic medical illnesses seen in ALL survivors in adulthood. Early interventions to restore the gut microbiome in ALL patients may potentially ameliorate the risk of long-term adverse health outcomes.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal